MetaPredict has been designed to get your drug discovery project more efficiently than ever before! It includes validated consensus models to predict a wide range of ADME-Tox properties. So what's new compared to other platforms? You can select only the most relevant models for your project and use custom thresholds to glow the molecular structures in accordance with your need.
Powerful
Smart platform
MetaPredict is a modeling platform which includes steps of sampling the chemical learning space, algorithms and descriptors in order to select the best conditions to predict the desired property. All models are validated, both internally and externally, with a relevant applicability domain. Finally, non-redundant models are used to create consensus models.
Intuitive
Simple API
We wanted to offer an interface to make our knowledge and predictive ADME-Tox models accessible to everyone. This API offers you in a few clicks the ability to predict one or more molecules, choose the models that are essential to your drug discovery project, and finally select the right thresholds to glow molecular structures according to your need.
Helpful
Revolutionary activity mapping
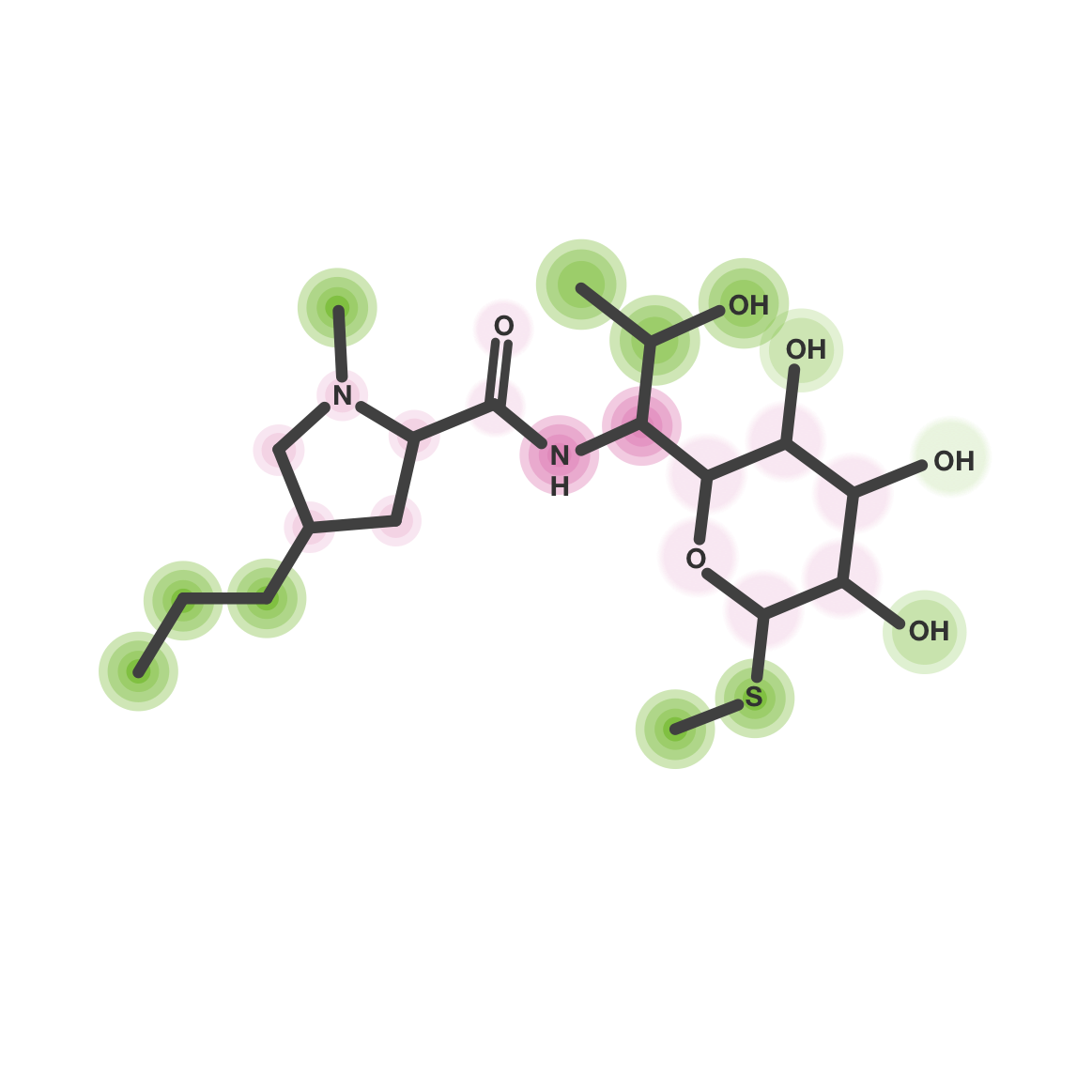
Our glowing molecules protocol is based on SAR grid generation to predict each simple and combined chemical substitutions. This takes into account local reliability and applicability domain of models in order to obtain the importance of each fragment for a specific chemical structure and glow only reliable information for the studied ADME-Tox property.
Inputs section
You can paste a SMILES/InChI, load a file or draw molecules. A single file in cvs, smi or sdf format is allowed with a maximum size of 1MB and 50 molecular structures. Once your structure or file has been transmitted, it is essential to click on the Upload Inputs button to start the molecular structures verification by the platform. If no error messages are displayed, you can go to the Models section. IMPORTANT NOTE: Only one file can be loaded at a time. If you upload new files, the previous one will be automatically deleted.
Models section
MetaPredict models are able to predict your molecules for the desired properties, but also offer activity maps that allow you to represent the selected properties on your molecular structures taking into account your own activity thresholds. To do this, you can use the Models section which contains:
Select: Select or deselect models.
Property: Model names and additional information ().
Threshold: Activity threshold for the selected property. For classification models, you can choose active category. For regression models, you can specify one or two activity thresholds (e. g. '> -4' or '> -4 < 0'). This information will condition the glowing molecules according to your active thresholds (green colour assigned to the fragments considered as active), as well as the calculation of the ADME-Tox score in the results table.
Highlight: Select or deselect activity map. If a model is selected and not this information, MetaPredict will only give you the model prediction for your molecules.
Submit
Once everything has gone smoothly, your molecules have been checked and your models have been selected, no errors () must be present. A button Submit must have made appear at the bottom of the page. You can now send your request to our server.