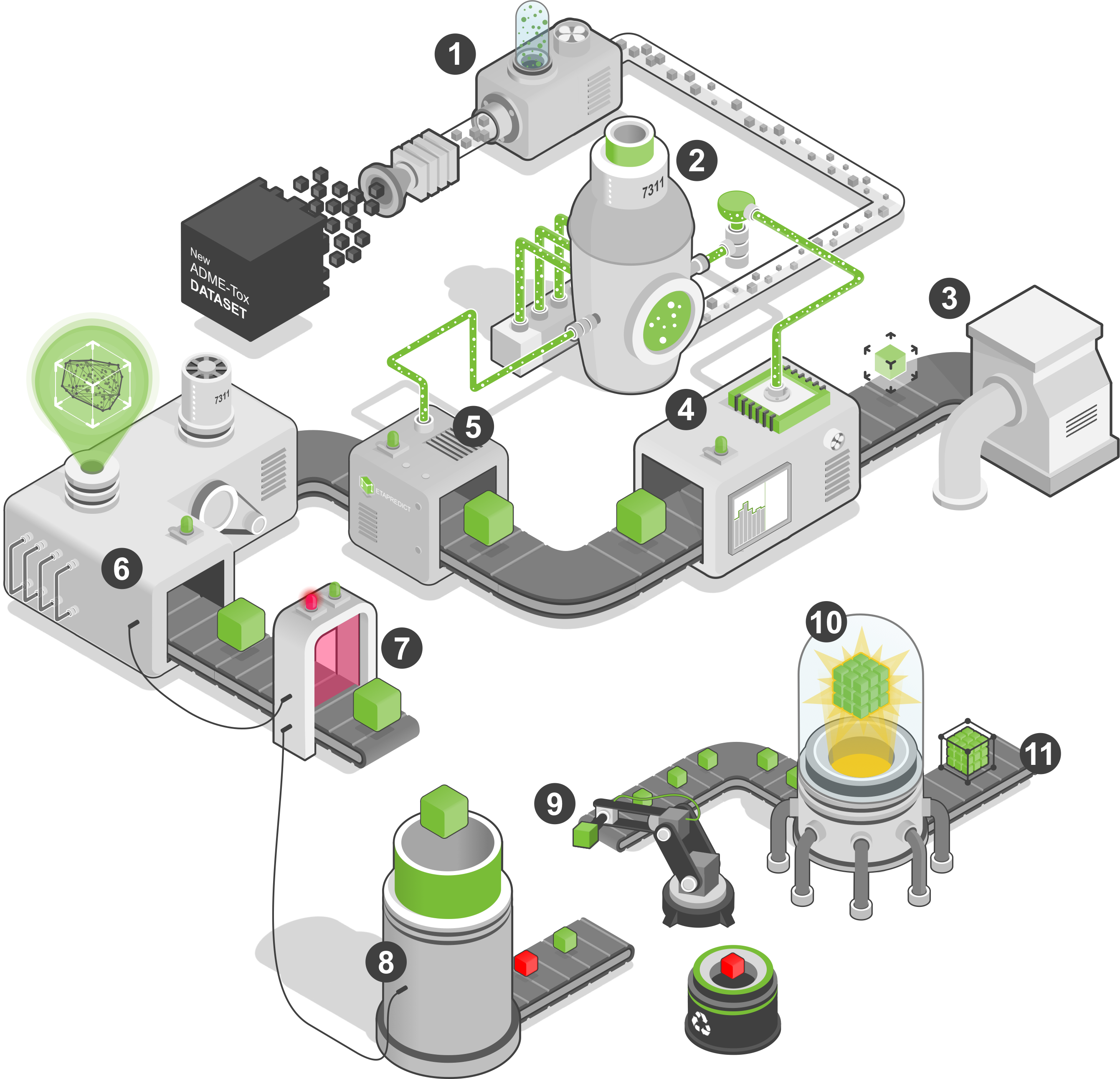
MetaPredict is a modeling platform with a particular focus on adherence to OECD QSAR best practices. First, the molecules are standardized by converting the structures into isomeric SMILES. At this stage the salts, solvents and fragments will be removed and the majority tautomer of each compound is then extracted. This preparation stage concludes with the generation of 2D or 3D structures of compounds and a structures ionization at a given pH using ChemAxon (1).
After normalization, the duplicates will be removed when two molecules have the same InChI Key are detected. This step consists of combining multiple bioactivity values for the same molecule (detection of potential univariate outliers) using an average within a regression model. Under a classification model, if a molecule has several non-conformant categories, the compound is automatically rejected from the data set. Molecular descriptors will be calculated for dataset without duplicates. Today, MetaPredict is able to calculate descriptors from RDKit, CDK, PaDEL, MORDRED, ISIDA, VolSurf+, as well as all fingerprints currently used by the scientific community. In addition, it is also able to use descriptors from tools external to these protocols. At the end of this step, an external dataset will be selected using a structural clustering where ~ 30% of the molecules of the dataset will be selected as external set (2).
The resulting modelling set (~70% of initial dataset) will undergo several random cuts in training and validation set (~75/25 respectively). the cutting of the data set is validated when the overlap of the chemical spaces of the training and test set is greater than 90% and that the averages of the biological activity are equal to the alpha risk of 5%. For each of these successive cuts, several models will be constructed by varying the algorithm and the descriptors used (3 and 4). When creating a simple QSAR model, several steps are performed. The first is to pre-process the data by removing zero variance descriptors and missing values. Then, as part of a regression model, the platform will test different correlation thresholds between the descriptors and the bioactive data points. For a classification, statistical average equality tests will allow to determine the most discriminating descriptors with regard to the properties studied (capable of separating the studied categories). The threshold for which the model performance (prediction of the training set and cross validation 5 folds) is optimal will be selected. This reduces the number of descriptors while keeping the most relevant to the property under study. Among the remaining descriptors, the most correlated ones will be deleted using the same exploratory approach. The creation of the model is finalized by a top-down stepwise step to keep only the most relevant descriptors in the model.
Validation and external sets are employed to determine the predictive power of the model (5). This precedes the creation of applicability domain that is performed using only with the learning and validation sets (6). Our applicability domain is based on a K-NN method. The most important descriptors (importance > 10%) will be used to create the Cartesian space. The values of the descriptors are weighted according to their own importance on the model. This allows us to reduce the impact of descriptors considered not important in the model to calculate an adjusted Euclidean distance. From the Cartesian space, we calculate the average distance of each individual from the training set with its nearest neighbours. An overall average distance is computed that will allow us to recalculate local distances for each individual based on the neighbors with a distance below this threshold. Each training individual will therefore have a specific threshold. This specific threshold is then weighted according to local reliability of each compound. The number of neighbors k is selected when the model's performance on the prediction of the validation set is optimal.
At the end, the model is considered valid if its performance is acceptable according to the threshold stated in the literature. A regression model is validated if its R2 is greater than 0.6, if its Q2 is greater than 0.6, if its R2f is greater than 0.5 and if the Golbraikh and Tropsha's criteria is less than 0.10. A classification model is validated if its MCC (Matthews correlation coefficient) is greater than 0.6, its MCCcv is greater than 0.6 and its MCCf is greater than 0.5 (7).
Only valid models will then be compared to generate the consensus model. Our consensus approach takes into account one step of valid models according to three criteria (8), namely the comparison of the chemical learning spaces covered, the comparison of the descriptors used and the comparison of the responses obtained by the models. The comparison of the chemical spaces covered is done by calculating the ratio of common molecules between the trainings set of all models. The comparison of descriptors is performed by comparing the descriptors that have been selected by each model according to the name of the descriptors and their order of importance. Finally, the comparison of predictions is made by calculating the correlation coefficient between the predicted values of each model. After combining these three pieces of information, we obtain a similarity coefficient for our models, which allows us to determine whether a model with poorer performance is already described by a higher performance model (9). At the end, ordered models are added successively to the consensus model. In this step, a model is rejected if it increases the prediction error. The consensus model is then finalized by providing it with all the procedures for preparing molecules, calculating descriptors, combining predictions and fields of applicability, as well as predicting new data sets. This step is completed by prediction of the external dataset in order to fully validate the consensus obtained (10).